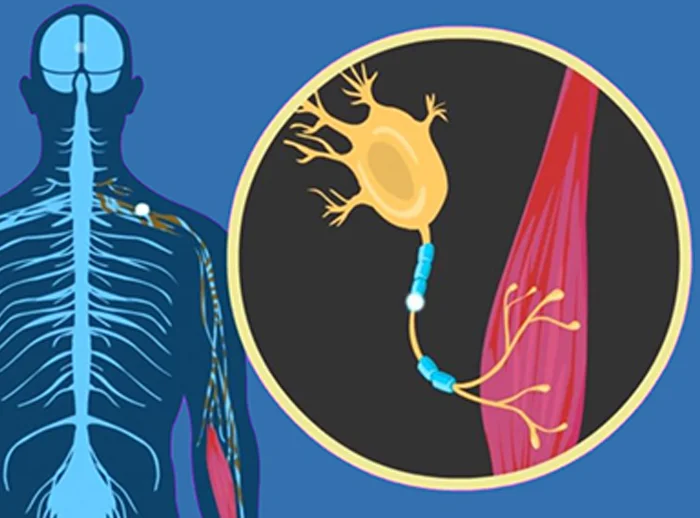
Amyotrophic Lateral Sclerosis (ALS)—also known as Lou Gehrig’s disease—is a progressive neurodegenerative disorder that affects nerve cells in the brain and spinal cord, leading to the loss of muscle control.
Causes of amyotrophic lateral sclerosis
The causes of Amyotrophic Lateral Sclerosis (ALS) are not fully understood, but research points to a combination of genetic, environmental, and cellular factors. Here’s a breakdown of the known and suspected causes:
Genetic Causes of amyotrophic lateral sclerosis
About 5–10% of ALS cases are familial (inherited), passed down through families.
Key Gene Mutations:
- C9orf72: Most common genetic cause; leads to toxic protein build-up.
- SOD1 (Superoxide Dismutase 1): Causes abnormal protein aggregation that damages neurons.
- TARDBP and FUS: Involved in RNA processing; mutations disrupt normal neuron function.
Environmental & Lifestyle Factors for Amyotrophic lateral sclerosis
Though most ALS cases are sporadic (90–95%), several environmental triggers are suspected:
- Toxins: Exposure to heavy metals (like lead), pesticides, or other neurotoxins.
- Military service: Higher rates of ALS have been observed in veterans—possibly due to chemical exposure or physical stress.
- Physical trauma: Some evidence suggests a link with head injuries.
- Exercise: High levels of strenuous physical activity have been debated as a potential risk.
- Infections: Certain viruses may contribute to neuronal damage (though evidence is limited).
Cellular and Molecular Mechanisms
Even in sporadic ALS, scientists have identified common dysfunctional processes, such as:
- Protein aggregation: Misfolded proteins accumulate and damage motor neurons.
- Glutamate toxicity: Excess glutamate overstimulates neurons, causing cell death.
- Oxidative stress: Imbalance between free radicals and antioxidants damages cells.
- Mitochondrial dysfunction: Impaired energy production leads to neuron death.
- Neuroinflammation: Chronic inflammation in the nervous system may accelerate progression.
amyotrophic lateral sclerosis Symptoms:
- Muscle weakness or stiffness
- Twitching (fasciculations), especially in arms and legs
- Slurred speech or difficulty swallowing
- Progressive muscle wasting
- Eventually leads to paralysis and respiratory failure
Types of ALS:
- Sporadic ALS: Most common form (~90–95%), occurs randomly without known family history.
- Familial ALS: Inherited form (~5–10%), associated with genetic mutations (e.g., SOD1, C9orf72).
Diagnosis:
No single test; diagnosis is clinical, supported by:
- EMG (electromyography)
- MRI
- Blood and urine tests (to rule out other diseases)
- Genetic testing (if family history is suspected)
Physiotherapy management of amyotrophic lateral sclerosis
Physiotherapy management plays a vital role in maintaining function, mobility, and quality of life in people with Amyotrophic Lateral Sclerosis (ALS). While physiotherapy cannot stop disease progression, it can help slow functional decline, reduce complications, and promote independence for as long as possible.
Goals
Maintain mobility and muscle strength
Prevent joint contractures and stiffness
Improve respiratory function
Reduce pain and fatigue
Maximize independence and safety
Educate patients and caregivers
Phases of Physiotherapy (Based on Disease Stage):
1. Early Stage ALS
- Mild weakness, minimal functional limitations
Management:
- Stretching exercises to prevent tightness (esp. in shoulders, calves)
- Range of motion (ROM) exercises
- Strengthening (gentle, non-fatiguing) of unaffected muscles
- Aerobic exercise (e.g., walking, swimming) as tolerated
- Fall prevention strategies
2. Middle Stage ALS
- Increased weakness, reduced mobility, risk of falls
Management:
- Use of orthotic devices (e.g., AFOs for foot drop)
- Assistive devices: canes, walkers, wheelchairs
- Continued ROM and stretching
- Postural correction to prevent scoliosis and pain
- Energy conservation techniques
3. Late Stage ALS
- Severe weakness, possible paralysis, wheelchair or bed-bound
Management:
- Passive ROM exercises to prevent joint stiffness
- Positioning techniques to prevent pressure sores
- Chest physiotherapy: postural drainage, assisted coughing
- Education on breathing techniques and secretion clearance
Collaboration with occupational therapists for transfer and mobility support.
What are the two main types of ALS, and how do they differ?
The two main types are sporadic ALS (90–95% of cases, with no family history) and familial ALS (5–10% of cases, inherited and linked to specific gene mutations like SOD1 and C9orf72).
What are common symptoms of ALS?
Common symptoms include muscle weakness or stiffness, twitching, slurred speech, difficulty swallowing, and progressive muscle wasting leading to paralysis and respiratory failure.
How does physiotherapy help manage ALS across its stages?
Physiotherapy helps by maintaining mobility, reducing pain and fatigue, and preventing complications. Management varies by stage, including stretching and strengthening in early ALS, assistive devices in the middle stage, and positioning and respiratory support in late-stage ALS.